Focus on identifying C3G and primary IC-MPGN
 Actor portrayal.
Actor portrayal.
The progressive, debilitating nature of 2 rare glomerular diseases
Originally defined as MPGN types I and III, IC-MPGN may be primary (idiopathic) or secondary (caused by other diseases and/or infections).4,6
80.9% of C3G patients and 33.5% of IC-MPGN patients had disease onset in childhood, per findings from the European Rare Kidney Disease Registry. Median (interquartile range) age at diagnosis was 12.4 (8.4-16.2) years for C3G and 38.9 (12.1-57.6) years for IC-MPGN.7
Across the US,
~5000 people are affected by C3G and primary IC-MPGN8
Clinical presentation of C3G and primary IC-MPGN is similar and characterized by complement overactivation leading to progressive kidney damage.2,9,10 The clinical signs and symptoms vary but can include:
-

Proteinuria2,9
-

Hematuria2,9
-

Decreased eGFR2
-

Hypertension2
-

Hyperlipidemia11
-

Edema/swelling12
-

Fatigue12
-

Drusen13,14
-

Low serum C315-17
-

Anxiety/depression12
Clinical evaluation is the first step in the diagnostic process, which also includes laboratory testing, renal biopsy and histology, and, in some cases, genetic testing.2,6,18
1
Initial clinical presentation
2
Serology* and urinalysis
3
Kidney biopsy
4
Immunological tests†
5
Genetic tests
Renal biopsy needed for definitive diagnosis
C3G and primary IC-MPGN have much in common, including clinical features, underlying causes, and how the diseases progress; however, they are considered distinct diseases.19,20
Renal biopsy is necessary to make a definitive diagnosis and differentiate between C3G and primary IC-MPGN. Biopsy samples are analyzed via light microscopy, immunofluorescence, and electron microscopy.2,9
Light microscopy
Provides initial diagnostic evaluation, assessing patterns of glomerular injury. Does not distinguish between forms of glomerulonephritis2,21
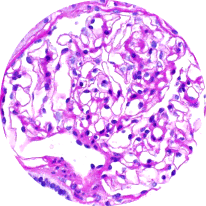
Healthy glomeruli
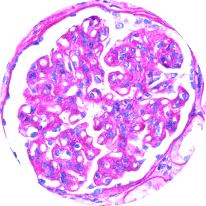
MPGN pattern of injury
(C3G/IC-MPGN)
Immunofluorescence
Distinguishes between C3G and primary IC-MPGN, detecting glomerular deposition of C3 through staining of C3 fragments and/or Ig2,6,21
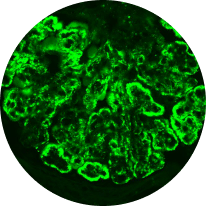
C3 deposits
(C3G)
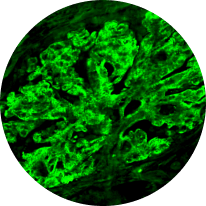
C3/Ig deposits
(IC-MPGN)
Electron microscopy
Distinguishes between C3G subtypes: dense deposit disease (DDD) and C3 glomerulonephritis (C3GN). Of the 2, DDD typically progresses more rapidly2,22
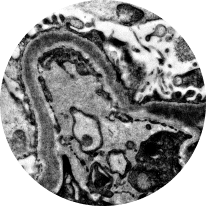
DDD
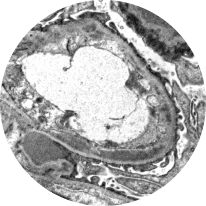
C3GN
C3G vs primary IC-MPGN under immunofluorescence
Histological analyses uncover disease activity and kidney damage in C3G and primary IC-MPGN. Under immunofluorescence, C3 staining intensity indicates the level of deposition of C3 breakdown products, providing diagnostic confirmation of C3G.2,23
Histological evidence from patients with repeat biopsies has shown that immunofluorescent staining patterns may change over time and sometimes lead to a switch from a diagnosis of primary IC-MPGN to C3G, or vice versa.23,24
C3G
C3G is defined by dominant glomerular C3 staining (≥2 orders of magnitude [OOM] compared with other immune reactants, including Ig).2-6
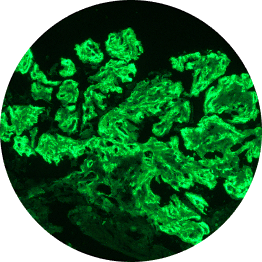

C3 dominant
Ig
Below are examples of C3 staining under immunofluorescence, with intensity ranging from 0 to 3+.
Increasing deposition
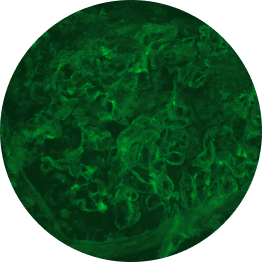
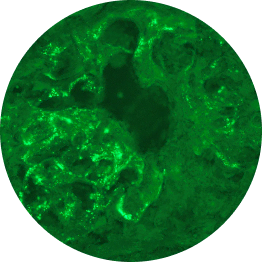
1+
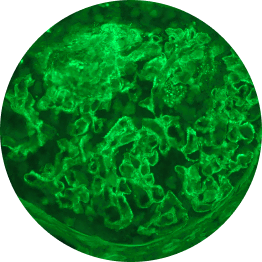
2+
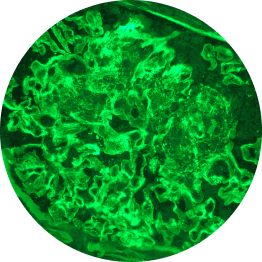
3+
1 OOM=10x; 2 OOM=100x.
Primary IC-MPGN
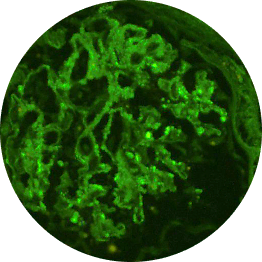
IC-MPGN exhibits Ig-dominant or codominant C3 deposition under immunofluorescent staining.4,6
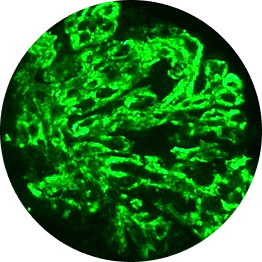
Ig
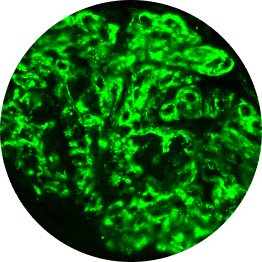
C3
Microscopic images: Courtesy of Patrick D. Walker, MD, Senior Renal Pathologist at Arkana Laboratories.
With the recent reclassification and new ICD-10-CM codes for these diseases and subtypes, specificity and accuracy support appropriate care management for your patients.25,26
N00.A – Acute nephritic syndrome with C3GN
N00.6 – Acute nephritic syndrome with DDD
N03.A – Chronic nephritic syndrome with C3GN
N03.6 – Chronic nephritic syndrome with DDD
N07.A – Hereditary nephropathy, not elsewhere classified with C3GN
N07.6 – Hereditary nephropathy, not elsewhere classified with DDD
N06.A – Isolated proteinuria with C3GN
N06.6 – Isolated proteinuria with DDD
N04.A – Nephrotic syndrome with C3GN
N04.6 – Nephrotic syndrome with DDD
N01.A – Rapidly progressive nephritic syndrome with C3GN
N01.6 – Rapidly progressive nephritic syndrome with DDD
N02.A – Recurrent and persistent hematuria with C3GN
N02.6 – Recurrent and persistent hematuria with DDD
N05.A – Unspecified nephritic syndrome with C3GN
N05.6 – Unspecified nephritic syndrome with DDD
N00.5 – Acute nephritic syndrome with diffuse mesangiocapillary glomerulonephritis
N03.5 – Chronic nephritic syndrome with diffuse mesangiocapillary glomerulonephritis
N07.5 – Hereditary nephropathy, not elsewhere classified with diffuse mesangiocapillary glomerulonephritis
N06.5 – Isolated proteinuria with diffuse mesangiocapillary glomerulonephritis
N04.5 – Nephrotic syndrome with diffuse mesangiocapillary glomerulonephritis
N01.5 – Rapidly progressive nephritic syndrome with diffuse mesangiocapillary glomerulonephritis
N02.5 – Recurrent and persistent hematuria with diffuse mesangiocapillary glomerulonephritis
N05.5 – Unspecified nephritic syndrome with diffuse mesangiocapillary glomerulonephritis

AH50=50% alternative hemolytic complement pathway activity; ANA=antinuclear antibodies; ANCA=antineutrophil cytoplasmic antibodies; CBC=complete blood count; C3G=C3 glomerulopathy; CH50=50% classical hemolytic complement pathway activity; eGFR=estimated glomerular filtration rate; ICD-10-CM=International Classification of Diseases, Tenth Revision, Clinical Modification; IC-MPGN=immune complex membranoproliferative glomerulonephritis; Ig=immunoglobulin; UPCR=urine protein-to-creatinine ratio.
References: 1. Donadelli R, Pulieri P, Piras R, et al. Unraveling the molecular mechanisms underlying complement dysregulation by nephritic factors in C3G and IC-MPGN. Front Immunol. 2018;9:2329. 2. Smith RJH, Appel GB, Blom AM, et al. C3 glomerulopathy—understanding a rare complement-driven renal disease. Nat Rev Nephrol. 2019;15(3):129-143. 3. Caravaca-Fontán F, Lucientes L, Cavero T, Praga M. Update on C3 glomerulopathy: a complement-mediated disease. Nephron. 2020;144(6):272-280. 4. Mastrangelo A, Serafinelli J, Giani M, Montini G. Clinical and pathophysiological insights into immunological mediated glomerular diseases in childhood. Front Pediatr. 2020;8:205. 5. Fakhouri F, Le Quintrec M, Frémeaux-Bacchi V. Practical management of C3 glomerulopathy and Ig-mediated MPGN: facts and uncertainties. Kidney Int. 2020;98(5):1135-1148. 6. Sethi S, Fervenza FC. Membranoproliferative glomerulonephritis—a new look at an old entity. N Engl J Med. 2012;366(12):1119-1131. 7. Schaefer F, Hofstetter J, Ruiz EM, et al. C3G and ic-MPGN across the life span: findings from the European Rare Kidney Disease Registry. Nephrol Dial Transplant. 2024;39(suppl 1). 8. Data on file. Apellis Pharmaceuticals, Inc., Waltham, MA. 9. Schena FP, Esposito P, Rossini M. A narrative review on C3 glomerulopathy: a rare renal disease. Int J Mol Sci. 2020;21(2):525. 10. National Kidney Foundation. 10 signs you may have kidney disease. Accessed November 6, 2024. https://www.kidney.org/news-stories/10-signs-you-may-have-kidney-disease 11. Rosenstein K, Tannock LR. Dyslipidemia in chronic kidney disease. In: Feingold KR, Anawalt B, Blackman MR, et al, eds. Endotext. South Dartmouth (MA): MDText.com, Inc.; 2022. 12. National Kidney Foundation. Voice of the patient. Accessed November 6, 2024. https://www.kidney.org/sites/default/files/C3G_EL-PFDD_VoP-Report_3-29-18.pdf 13. American Academy of Ophthalmology. What are drusen? Accessed November 6, 2024. https://www.aao.org/eye-health/diseases/what-are-drusen 14. Heiderscheit AK, Hauer JJ, Smith RJH. C3 glomerulopathy: understanding an ultra-rare complement-mediated renal disease. Am J Med Genet C Semin Med Genet. 2022;190(3):344-357. 15. Medjeral-Thomas NR, O’Shaughnessy MM, O’Regan JA, et al. C3 glomerulopathy: clinicopathologic features and predictors of outcome. Clin J Am Soc Nephrol. 2014;9(1):46-53. 16. Pickering MC, D’Agati VD, Nester CM, et al. C3 Glomerulopathy: consensus report. Kidney Int. 2013;84(6):1079-1089. 17. Zhang Y, Nester CM, Martin B, et al. Defining the complement biomarker profile of C3 glomerulopathy. Clin J Am Soc Nephrol. 2014;9(11):1876-1882. 18. Lafayette RA, Charu V. Expert discussion on challenges in C3G diagnosis: a podcast article on best practices in kidney biopsies. Adv Ther. 2023;40(12):5557-5566. 19. Noris M, Daina E, Remuzzi G. Membranoproliferative glomerulonephritis: no longer the same disease and may need very different treatment. Nephrol Dial Transplant. 2023;38(2):283-290. 20. Cook HT. Evolving complexity of complement-related diseases: C3 glomerulopathy and atypical haemolytic uremic syndrome. Curr Opin Nephrol Hypertens. 2018;27(3):165-170. 21. Kidney disease: improving global outcomes (KDIGO) glomerular diseases work group. KDIGO 2021 Clinical practice guideline for the management of glomerular diseases. Kidney Int. 2021;100(4S):S1-S276. 22. Bomback AS, Santoriello D, Avasare RS, et al. C3 glomerulonephritis and dense deposit disease share a similar disease course in a large United States cohort of patients with C3 glomerulopathy. Kidney Int. 2018;93(4):977-985. 23. Hou J, Markowitz GS, Bomback AS, et al. Toward a working definition of C3 glomerulopathy by immunofluorescence. Kidney Int. 2014;85(2):450-456. 24. Daina E, Cortinovis M, Remuzzi G. Kidney diseases. Immunol Rev. 2023;313:239-261. 25. Centers for Disease Control and Prevention. ICD10 Coordination and Maintenance Committee Meeting Diagnosis Agenda. March 5-6, 2019, Part 2. Accessed November 6, 2024. https://archive.cdc.gov/www_cdc_gov/nchs/data/icd/Topic-packet-March-2019-Part-2Vs3.pdf 26. Centers for Disease Control and Prevention. National Center for Health Statistics—ICD-10-CM. Accessed November 6, 2024. https://icd10cmtool.cdc.gov/?fy=FY2024&query=immune%20complex